맥레오드증후군(McLeod syndrome)은 다기관을 침범하는 드문 X연관유전질환으로 가시세포증가무도병(choreoacanthocytosis)과 함께 대표적인 신경가시세포증가증(neuroacanthocytosis)의 한 형태이다. 대부분 40-50대의 나이에 무도병, 경련, 인지기능 저하, 정신병 등의 신경계증상이 시작되며 말초신경병증, 근육병증, 심근병증, 가시적혈구증가증과 간비장비대 등 다기관 침범이 동반된다. 현재까지 국내에는 보고된 사례가 없었으나, 저자는 유전자 분석을 통해 60세 한국인 남성을 맥레오드증후군으로 진단한 증례를 경험하여 이를 보고하고자 한다. 환자는 XK유전자분석에서 이전에 보고된 바 있는[1] 엑손3의 5개 염기서열의 결실(c.856-860del-CTCTA)이 확인되었다.
증 례
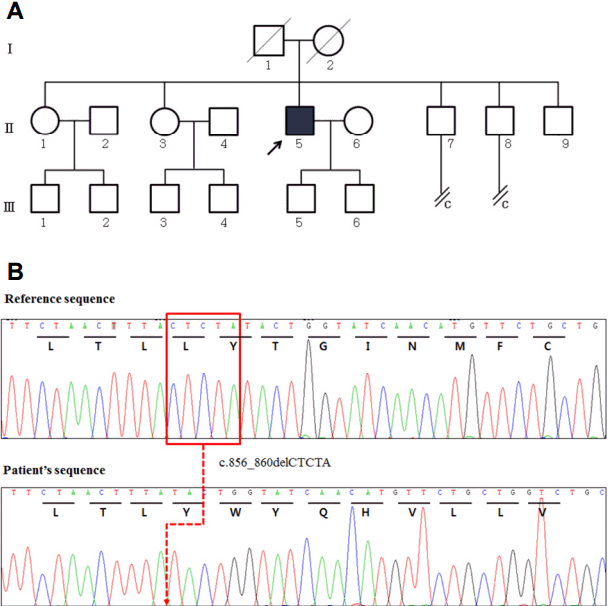
60세 남자(Ⅱ-5, Fig. 1-A)가 7년 전부터 점차 진행하는 무도병을 주소로 내원하였다. 7년 전부터 불수의적으로 혀를 날름거리며 음식물을 흘리기 시작하였는데, 점차 사지와 몸통에도 불수의적인 움직임이 시작되었다. 환자는 20년 전부터 원인미상의 간부전에 의한 식도정맥류로 토혈이 수 차례 반복되어 내시경적 정맥류 결찰을 시행 받은 바 있었다. 가족력에서 환자의 막내 남동생(Ⅱ-9, Fig. 1-A)이 약 20년 전 두부 수상 후 정신질환이 발생한 뒤 현재까지 연락이 닿지 않는다고 하였다. 다른 형제들은 특별한 병력이 없었다.
내원 당시 환자는 혀를 내밀고 유지하거나, 팔, 다리를 특정 자세로 유지하는 것이 불가능한 운동지속불능증(motor impersistence)을 보였다. 혀를 반복적으로 날름거리며 사지를 불규칙적으로 짧게 비틀거나 꼬는 움직임이 지속적으로 관찰되었다. 이는 상지보다 하지에서 두드러졌고, 수초 정도는 의식적인 억제가 가능하였다. 떨림이나 운동완만은 관찰되지 않았다. 대졸 학력으로 간이정신상태검사는 30점이었으며, 뇌신경기능장애는 없었다. 상하지의 근력은 정상이었으나 근위부 근육의 위축이 관찰되었고 심부건반사가 나타나지 않았다. 상하지 원위부에서 진동감각과 위치감각이 중등도 이상 감소하여 있었으며 Romberg징후 양성 소견을 보였다.
시행한 실험실 검사에서 범혈구감소증이 확인되었고, 말초혈액펴바른표본에서 가시적혈구(acanthocyte)가 4% 가량 관찰되었다. 환자의 혈액과 식염수를 1:1로 섞어 도말한 뒤 위상차현미경으로 관찰하였을 때, 가시적혈구의 수가 매우 증가함을 확인할 수 있었다. 또한 혈청 크레아틴키나아제(creatine kinase, CK), 젖산탈수소효소와 미오글로빈의 혈중 농도가 각각 803 IU/L, 378 IU/L와 221 ng/mL로 증가하여 있었다. 신경전도검사에서 양측 정중신경, 비골신경과 경골신경의 복합근활동전위(compound muscle action potential, CMAP)가 감소되었고, 표재비골신경의 감각신경활동전위(sensory nerve action potential, SNAP)는 측정 불가능하였으며 양측 정중신경, 척골신경과 비복신경의 SNAP가 감소하여 있었다. 즉 가시적혈구증가증, 근육병증과 말초신경병증이 함께 존재함을 확인하였다.
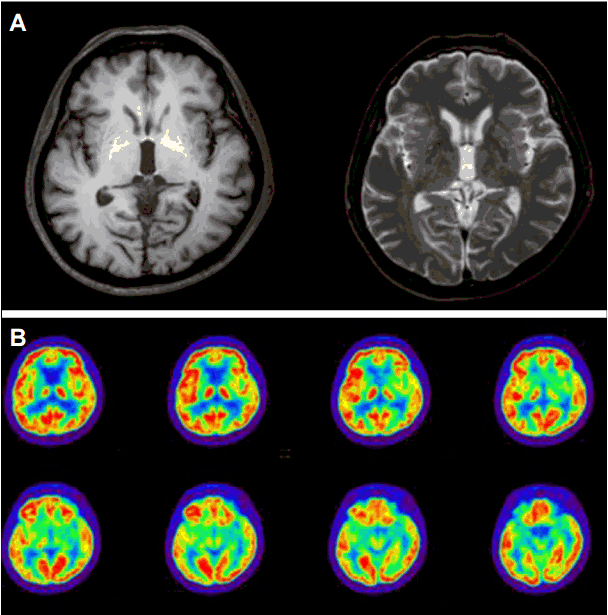
뇌MRI (brain magnetic resonance imaging)에서는 양측 꼬리핵 및 조가비핵의 부피 감소가 관찰되었고, 양측 창백핵의 T1강조영상에서의 신호강도 상승이 확인되었다(Fig. 2-A). [18F]FP-CIT 양전자방출단층촬영([18F]FP-CIT PET) 영상에서 양측 꼬리핵의 도파민전달체결합(dopamine transporter binding)이 경미하게 감소해 있었다. 뇌 [18F]FDG양전자방출단층촬영([18F]FDG-PET)에서 양측 꼬리핵과 조가비핵의 대사저하소견이 관찰되었다(Fig. 2-B).
감별진단을 위해 시행한 치아적핵담창구루이체위축증(dentatorubropallidoluysian atrophy, DRPLA)와 헌팅턴병에 대한 유전자검사에서 각각 12번 염색체와 4번 염색체 단완의 CAG반복수는 정상이었다. 혈청 구리와 세룰로플라스민 농도 역시 정상이었다.
이를 종합한 감별진단으로, 늦은 나이에 발생하여 다기관 침범이 동반된 neuroacanthocytosis라는 점에서 맥레오드증후군과 choreoacanthocytosis를 고려하게 되었다. 중년의 나이의 발병하여 근육병증과 간부전이 동반된 반면 교순벽(lip biting), 교설벽(tongue biting)이나 근긴장증 등이 동반되지 않은 점 등은 choreoacanthocytosis보다 멕레오드증후군을 시사한다. 이에 따라 저자들은 XK유전자에 대한 Sanger염기서열분석을 시행하였다. 그 결과 환자의 XK유전자의 엑손3의 856번부터 860번 5개의 뉴클레오티드 CTCTA (c.856_860delCTCTA)의 결실이 확인되었다. 이 결실은 프레임시프트를 일으켜 후속의 16번째 코돈에서 조기의 종결 코돈(TGA)을 형성한다(p.Leu286Tyrfs*16) (Fig. 1-B). 이는 2001년 Danek 등[1]에 의해 보고된 된 돌연변이로, 본 환자를 맥레오드증후군으로 진단하였다.
고 찰
맥레오드증후군은 choreoacanthocytosis와 함께 neuroacanthocytosis증후군의 핵심을 이루는 질환이다. Neuroacanthocytosis증후군은 적혈구 표면에 돌출된 돌기를 가지는 가시적혈구의 증가와 신경계통의 이상을 함께 보이는 질환군을 일컫는다. Choreoacanthocytosis는 보통염색체열성의 형태로 유전되며 전세계적으로 1,000명 가량의 환자가 보고된 반면, 맥레오드증후군은 X연관열성유전(X-linked recessive inheritance, XR)의 형태로 유전되며 현재까지 150명 가량의 증례가 보고되었다[2]. 아시아에서는 일본, 중국[3]과 대만[4] 등지에서 보고되었으나 현재까지 국내에서는 보고된 바 없어 한국에서 첫 번째 증례라는 의미가 있다.
맥레오드증후군의 신경계증상은 다른 유전운동질환과 비교하여 늦게 발생하는 편으로 대개 40-50대에 시작된다. 그 중 무도병은 대부분의 환자에서 발생하는데, 무도병의 양상만으로 헌팅턴병 등의 기타 질환과 감별하기는 어렵다. 단, choreoacanthocytosis에 비해 교순벽, 교설벽, 삼킴장애, 근긴장이상증이나 파킨슨증은 매우 드물다는 점이 감별에 도움이 된다[5]. 우울증, 정신분열증, 강박장애 등의 정신질환이 운동질환에 선행할 수 있다. 환자의 막내 남동생이 20대에 정신질환이 발생한 후 연락이 두절되었기에 감별진단에 도움이 되리라 예상하여 연락을 요청하였으나 환자의 의사에 반하여 시행할 수 없었다. 질병의 후기에는 인지기능 저하가 동반될 수 있고, 환자의 약 반수에서 전신발작(generalized seizure)이 병발한다. 또한 대부분의 환자에서 말초신경병증 및 CK의 혈중 농도 상승을 확인할 수 있다. 가시적혈구의 증가도 대부분의 환자에서 관찰되지만, 그것이 없거나 매우 적다고 하여 맥레오드증후군을 배제할 수 있는 것은 아니다. 그러한 경우에는 환자의 혈액과 식염수를 1:1로 섞어 위상차현미경으로 가시적혈구가 나타나거나 크게 증가함을 확인하는 것이 도움이 된다. 심근병증은 60%가량의 환자에서 동반된다. 간비장비대는 약 40%의 환자에서 발생하는데, 가시적혈구가 정상 적혈구에 비해 망상내피계(reticuloendothelial system)에서 빨리 제거되면서 혈관외용혈이 발생하여 발생한다고 추정하고 있다[6].
간부전을 앓아오던 환자에서 무도병이 발생하는 경우 유병율을 고려하여 간뇌변성(hepatocerebral degeneration)이나 윌슨병 등을 우선 감별해야 한다. 본 증례는 이들이 배제된 뒤 발병 연령과 다기관 침범의 양상을 고려하여 맥레오드증후군에 대한 유전자검사를 시행하게 된 증례이다. 환자는 반복되는 토혈로 간생검을 포함한 광범위한 검사를 시행하였으나 간부전의 다른 원인은 배제가 되었던 상태였다. 따라서 맥레오드증후군이 간부전의 원인이거나 적어도 크게 기여할 것으로 생각되는데, 그 정도가 매우 중증이었음이 주목할 만 하다. 맥레오드증후군에서 간비장비대는 대개 양성의 경과를 보이나 드물게 심각한 간기능저하를 보이기도 한다. 반면 choreoacanthocytosis에는 11%가량의 환자에서 간비대가 동반되나 중증의 간부전은 보고된 바 없다[7]. 이 역시 본 환자에서 choreoacanthocytosis보다 맥레오드증후군을 시사하는 실마리이다. 환자와 동일한 유전자형을 보였던 증례 보고들에서는 간부전이 관찰되지 않았는데[1,3], 이는 같은 유전자형이라도 그 표현형은 다를 수 있음을 시사한다.
맥레오드증후군은 XK유전자의 돌연변이로 인해 발생한다. X 염색체 단완에 위치한 이 유전자는 XK단백질을 발현한다. Kell혈액형 시스템은 ABO, Rh 다음으로 강력한 혈액형 시스템인데, 바로 XK단백질이 적혈구막에서 Kell단백질과 결합하여 Kx항원을 발현하는 역할을 한다. 따라서 맥레오드증후군에서 적혈구의 Kx항원과 Kell단백질의 발현이 매우 감소하여 있는 것을 확인할 수 있다. 최근 맥레오드 형질에서 가시적혈구증가증이 발생하는 기전이 밝혀졌다. XK단백질-Kell단백질복합체가 적혈구 표면의 4.1단백질복합체의 일부를 이루는데, 맥레오드 형질에서는 4.1단백질-스펙트린-액틴 상호작용이 유지되지 못하여 적혈구막이 불안정해지고 가시적혈구증을 일으킨다는 것이다[8]. 반면 신경계증상의 발생기전은 정확히 알려져 있지 않다. 다만 Kell단백질이 엔도텔린(endothelin)의 합성 과정에 관여하는 것이 알려진 바가 있어[9] 엔도텔린수용체B (endothelin receptor B)가 뇌의 도파민 분비에 관여함을 고려할 때, 이것이 신경계증상 발생에 기여할 것으로 추정할 뿐이다.
진단은 적혈구의 Kx항원과 Kell단백질의 발현이 매우 감소해있는 것을 확인하거나, XK유전자검사를 통해 이루어진다. 본 환자에서는 XK유전자의 엑손3의 5개 염기서열의 결실이 확인되었다. 이 돌연변이는 2001년 최초로 보고되었으며[1], 최근 최초의 중국인 멕레오드증후군에서도 동일한 돌연변이가 확인되었다[3].
맥레오드증후군에 대한 치료는 대증적 치료를 시행한다. 무도병을 비롯한 운동과다증에 대하여 tetrabenazine 또는 비전형항정신병약제(quetiapine, clozapine 등)가 도움이 되며 환자의 약 반수에서 발생하는 경련발작도 단일 항경련제로 조절되는 편이다. 뇌심부자극술(deep brain stimulation)을 시행한 증례들에서는 그 결과가 일정하지 않아 이상적인 자극위치와 자극 변수들에 대한 연구가 더 필요한 실정이다[2]. 심근병증과 부정맥 등의 심장 합병증이 사망의 주 원인이므로 주기적인 모니터링이 필요하며, Kell혈액형 시스템과 관련하여 항원항체반응이 일어날 수 있으므로 수혈 시 주의가 필요하다. 본 환자의 경우도 식도정맥류 파열이 반복된다면 수혈이 불가피할 수 있으므로, 환자의 질환에 대한 정확한 이해가 필요하다.
본 증례는 한국인에서 유전자검사로 진단된 최초의 맥레오드증후군으로 그 의미가 있으며 향후 국내 neuroacanthocytosis에 대한 연구에 도움이 될 수 있을 것으로 생각한다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print