증 례
52세 여성이 내원 4개월 전부터 목가누기 및 삼키기가 불편해져 내원하였다. 처음에는 음식을 씹는 것이 불편하였으나 삼키는 것이 힘들어졌고 내원 당시에는 물만 마셔도 사래가 들린다고 호소하였다. 타 병원에서 내시경검사를 받았으나 이상은 없다고 하였다. 신경계진찰에서 목가누기가 Medical Research Council 척도 4점으로 악화되어 있었고 혀의 위축이 확인되었고, 경한 발음장애가 있었다. 이외 뇌신경검사, 감각검사 및 심부건 반사는 이상이 없었고 병적반사는 관찰되지 않았다. 발음장애 및 삼킴장애는 조금 쉬면 나아지나 큰 차이는 없다고 하였다. 가족력 및 과거력에서는 이상이 발견되지 않았다. 혈액검사도 면역 관련된 검사는 정상이었고 근육크리아티닌 수치는 216 IU/mL (정상: ~180 IU/mL)로 증가되어 있었다.
질문 1. 이 환자의 감별진단은?
환자는 병력에서 경한 발음장애도 있지만, 주로 진행하는 삼킴장애를 주소로 내원하였다. 삼킴장애는 내과적 질환을 포함하여 다양한 신경병, 신경근육접합부 및 근육질환에서 발생할 수 있다. 내과적 질환으로는 역류성식도염과 결합조직질환 등이 있을 수 있다. 신경의 질환으로는 운동신경을 침범하는 다양한 운동세포병과 길랑-바레증후군 등을 고려할 수 있다. 신경근육접합부 질환으로는 중증근무력증과 렘버트이튼증후군 등을 고려해야 한다. 근육질환으로도 다양한 유전성 근육병과 염증성 근육병도 고려되야 한다. 더 자세한 병력청취도 중요하다. 삼킴장애가 고형식일 때 발생하는지, 액체류에도 발생하는지 확인할 필요가 있다. 또 삼킴장애가 간헐적인지 지속적인 지도 중요한 병력청취가 될 수 있다. 환자가 복용 중인 약제에 대한 청취도 도움이 될 수 있다. 고형식에만 문제가 있을 경우 종양 혹은 협착 등에 의한 가능성이 높다. 반면 액체류 삼킴도 문제가 있다면 앞서 언급된 다양한 신경근육질환에 대한 감별이 필요하다. 오랜 기간의 약물 복용에 의한 식도염일 가능성도 있기 때문에 약물에 대한 조사도 놓치지 말아야 한다. 기능적으로 삼킴장애는 크게 해부학적으로 구강인두 및 식도 부위의 문제로 나뉠 수 있다[1]. 일반적으로 삼킴 자체가 힘들거나, 삼킴 이후에 기침 및 기도 역류 등의 문제가 있을 경우 식도보다는 그 상부인 구강인두의 삼킴장애일 가능성이 높다.
감별해야 할 질환은 Table 1에 기술된 바와 같다. 우선 운동세포신경병의 경우, 연수형 근육위축가쪽경화증(amyotrophic lateral sclerosis)의 경우 초반부터 발음장애와 삼킴장애가 올 수 있어 이에 대한 고민이 필요하다. 또한 남성의 경우에는 척수구근위축중(spinal and bulbar muscular atrophy) 또는 케네디병이라 불리는 유전성 운동세포신경병을 고려해야 한다. 이 질환 또한 초기부터 삼킴장애가 발생하기 때문이다. 하지만 면밀한 신경계진찰 만으로 감별이 어느 정도 가능하다. 근육위축가쪽경화증은 병적반사가 동반되어 있을 가능성이 높기 때문에 이에 대한 확인이 감별진단에 도움을 줄 수 있다. 케네디병의 경우, 이 질환에서 아주 초기에 나타날 수 있는 손떨림 그리고 얼굴근육 주위의 근연축이 특징적이므로 이에 대한 확인이 도움될 것으로 생각된다. 근육질환의 경우도 유전성 질환인 눈인두성근디스트로피(oculopharyngeal muscular dystrophy), 긴장성근디스트로피(myotonic dystrophy) 등에 대한 감별이 필요하며, 염증성 근육병인 면역매개성괴사성근육병(immune mediated necrotizing myopathy)과 봉입체근염(inclusion body myositis)의 경우에도 삼킴장애가 흔히 동반된다. 근육질환의 경우 일반적으로 삼킴과 관련된 근육뿐만이 아니라 다른 근육도 침범을 하기 때문에 이에 대한 면밀한 조사가 필요하다. 그리고 임상적으로 근긴장증이 동반되어 있는지도 확인하면 감별진단에 도움이 된다. 추가적으로 근육의 침범이 비대칭적인 형태를 보인다면 봉입체근염을 더 의심해 볼 수 있다. 신경근육접합부질환의 경우, 중증근무력증은 눈꺼풀 처짐, 복시 등의 동반증상의 여부 확인과 함께 일중 변동성이 있는지 확인하는 것이 도움된다. 렘버트이튼증후군의 경우에는 입마름과 같은 자율신경계 이상의 동반 여부를 추가적으로 확인할 필요가 있다.
질문 2. 진단에 필요한 검사는?
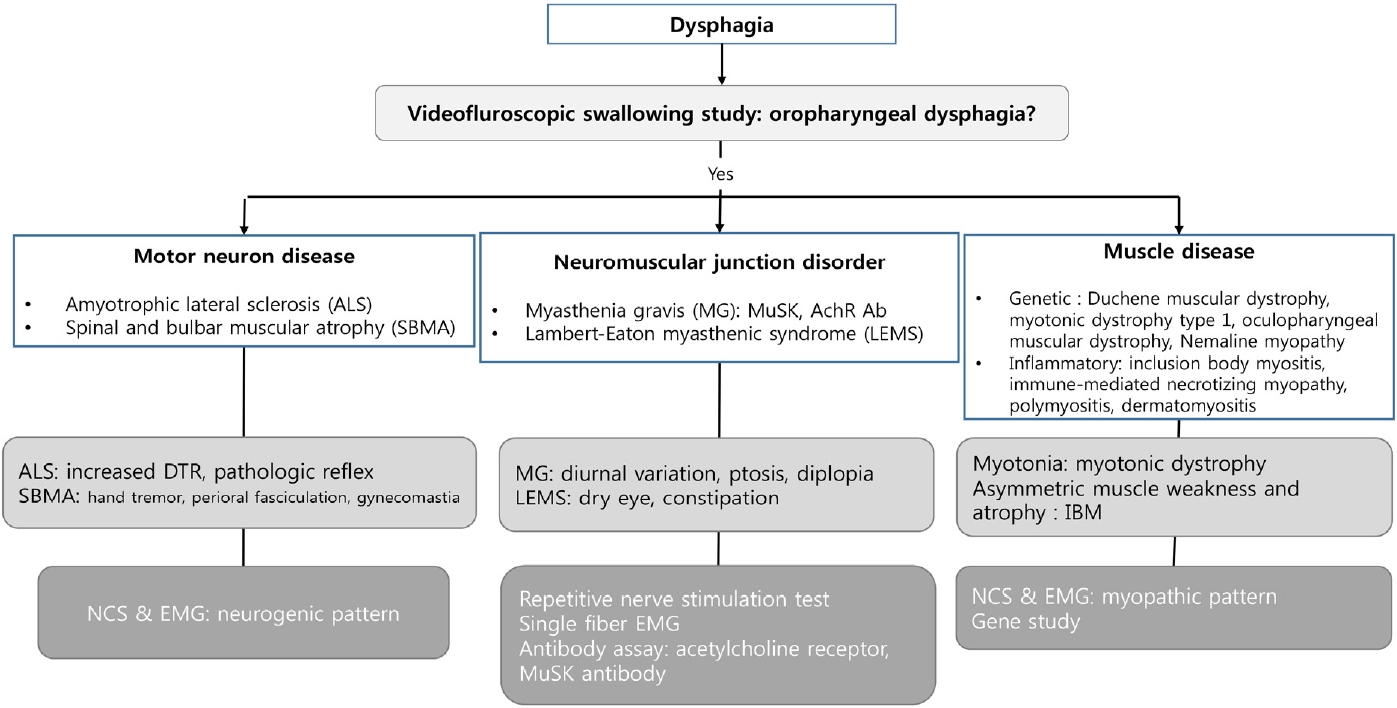
환자는 우선 객관적 검사를 통해 삼킴장애의 위치 및 상태에 대한 파악이 필요하다. 삼킴장애에 대한 검사는 일반적으로 비디오투시삼킴검사(videofluroscopic swallowing study)가 사용되나, 바리움 식도조영검사(barium esophagography), 코인두두경검사(nasopharngoscopy), 위내시경(esophagogastroduodenoscopy) 그리고 컴퓨터단층촬영(computed tomography) 등이 사용될 수 있다. 일반적으로 비디오투시삼킴검사가 구강인두 삼킴장애를 잘 반영하기 때문에 신경과에서는 이 검사를 주로 사용한다[2]. 비디오투시삼킴검사로 삼킴의 개시점, 음식물의 흡인(aspiration) 여부, 코인두의 역류(regurgitation) 그리고 삼킴 이후 인두에 남는 잔여량을 객관적으로 확인할 수 있다. 즉 비디오투시삼킴검사를 통해 구강인두의 삼킴장애인지, 식도 부위의 삼킴장애인지를 확인하여 문제점을 국소화하여 진단에 도움이 될 수 있다.
이를 통해 신경계질환의 문제로 의심이 되면 다양한 신경생리검사를 통해 감별이 더 가능하다. 운동신경의 이상 유무를 확인하기 위해 신경전도검사가 도움될 것이다. 이와 함께 침근전도검사를 시행해서 근육병의 가능성을 같이 확인할 수 있다. 연수형 근육위축가쪽경화증인 경우 혀 및 삼킴관련된 근육에서 초기에 침근전도 검사를 통해 비정상적인 탈신경 활동전위를 확인할 수 있다. 초기 근근육질환이라면 근육병성 패턴(myopathic pattern)의 침근전도 소견을 보일 것이며, 긴장성근디스트로피의 경우에는 근긴장방전(myotonic discharge)이 보일 것이다. 또한 신경근육접합부의 이상 여부를 확인하기 위해 반복자극신경검사나 단일근섬유근전도검사 등을 시행할 필요가 있다. 신경근육접합부 후시냅스의 장애인 경우 반복자극신경검사에서 저빈도 자극시 10% 이상의 감소가 확인될 수 있으며, 전시냅스의 장애인 경우 고빈도 자극시 100% 이상의 증가가 확인된다. 이러한 신경생리검사를 통해 어느 정도 질환에 대한 감별이 될 것으로 생각된다.
본 증례의 경우 저빈도 반복신경자극검사에서 10% 이상의 감소가 보여 신경생리학적으로는 중증근무력증에 합당하였다. 하지만 아세틸콜린항체검사에서는 음성이 확인되었다. 이후 muscle specific antibody (MuSK)항체검사를 시행하였고 양성(0.5 nmol/L)으로 확인되었다.
질문 3. 적절한 치료는?
본 증례는 MuSK 양성 전신형 중증근무력증으로 진단할 수 있었다. 이 환자의 Myasthenia Gravis Clinical Score (MGCS)는 12점이였고 스테로이드 치료를 시작하였으나 큰 호전이 없어 면역글로불린 2 g을 사용하였다. 임상적 호전을 보이며 MGCS 4점으로 퇴원을 하였으나 약 한 달 뒤 삼킴장애가 다시 악화되며 MGCS 14점으로 확인되었다. 난치성 전신중증근무력증으로 판단하여 리툭시맙을 이전 문헌과 같이 375 mg/m2를 매주 4회 투여 후 매달 한 차례씩 3회 투여하였다[3]. 이후 환자의 증상이 서서히 완화되며 현재는 프레드리솔론 10 mg과 아자티오프린 100 mg 복용하며 안정적으로 외래를 다니고 있다.
토 의
IgG1형의 면역글로불린을 형성하는 아세틸콜린수용체 항체와는 다르게 MuSK 항체는 IgG4형의 면역글로불린이며, 단백질의 기능을 직접적으로 차단시키는 것으로 알려져 있다. 최근 들어 low-density lipoprotein receptor-related protein 4 (LRP4)항체도 발견되며 발병 기전이 더 자세히 밝혀지게 되었다. 정상인에서는 MuSK-LRP4 사량체 복합체(tetrameric complex)가 형성이 되고이 복합체에 agrin과 함께 붙게 되면, 인산화반응을 통해 아세틸콜린수용체의 클러스터를 발현시키는 것으로 이해된다. 하지만 여기에 MuSK항체가 작용을 하면 사량체 복합체가 형성을 방해하여 아세틸콜린수용체의 클러스터가 군집을 이루지 못하고, 결국 신경 접합부의 신경전달에 문제가 생기는 것으로 추측하고 있다[4].
MuSK항체 관련 중증근무력증은 전체 중증근무력증 환자의 약 5-8%를 차지한다[5]. 특히 젊은 여성에게 많이 발생하는 것으로 알려져 있으며 흉선종과는 연관성이 없으며, HLA 타입과의 연관성이 제시되고 있다. 특징적으로 MuSK항체 관련 중증근무력증은 비교적 빠르게 진행하며 대부분의 경우 연하장애를 동반한 연수증상이 더 뚜렷한 전신형 중증근무력증으로 나타나는 경우가 많다[6]. 삼킴장애 이외에 비음장애와 저작기능에도 문제가 잘 생긴다. 특히 진행이 빠른 경우에는 중증근무력증 위기까지 오는 경우도 있다. 근력의 약화가 있는 경우에는 몸통의 축위주 근육 약화가 잘 생기며 목가누기가 두드러지게 약화된다. 특히 목가누기는 치료에도 효과가 미미한 경우도 많다. 또 하나의 임상적 특징은 아세틸콜린 항체 양성 중증근무력증에 비해 혀 근육의 위축이 잘 발생한다. 앞서 언급된 바와 같이 흉선종과의 연관성이 낮기 때문에 흉선 제거술이 효과적인 치료 방법은 아닌 것으로 알려져 있다[7].
고전적으로 중증근무력증은 신경계진찰 및 꼼꼼한 문진을 바탕으로 약물검사, 신경생리학적 검사를 통해 진단을 하였고, 항체 확인을 위한 혈액학적 검사도 도움이 되었다. 하지만 최근 연구들에 의하면 MuSK항체 관련 중증근무력증에서 에드로포니움 또는 피리도스티그민 약물검사에 양성을 보일 확률은 약 40-75%로, 아세틸콜린항체 양성 중증근무력증에 비해 낮은 것으로 알려져 있다. 반복신경자극검사도 아세틸콜린항체 양성 중증근무력증에 비해 양성률이 낮으며, 양성으로 나올 경우에도 근위부 근육군에서 확인되는 경우가 많다고 알려져 있다[8]. 하지만 최근 아세틸콜린 항체 외의 다른 여러 항체의 역할이 밝혀지면서, 중증근무력증을 항체와 연령에 따라 재분류하는 시도가 이루어지고 있다(Table 2) [9].
MuSK항체 관련 중증근무력증으로 진단된 경우 치료적 접근도 다르게 이루어져야 한다. MuSK항체에 의해 아세틸콜린 수용체의 발현이 되지 않는 것이 주된 병리 기전으로 이해되기에 메스티논과 같은 아세틸콜린분해억제제가 일반적으로 큰 도움이 되지 않는다. 현재까지는 스테로이드와 함께 다양한 면역억제제가 효과적이다. 약 10-15%의 MuSK항체 관련 중증근무력증이 난치성으로 진행하며 이러한 경우에는 빠른 면역글로불린 및 혈장교환술이 도움이 되며, 특히 MuSK항체 관련 중증근무력증에서 급성기 치료로는 면역글로불린보다 혈장교환술이 더 효과적이다는 보고도 있다[10]. 최근 3,4 diaminopyridine이 큰 부작용 없이 치료적으로 사용될 수 있다는 보고도 있으나[11], 더 많은 연구가 필요하다. 또 항CD20단클론키메라 항체인 리툭시맙이 난치성 중증근무력증 연구에서 의미 있는 치료효과를 보이고 있다[12]. 또 주목할 만한 것은 아세틸콜린항체 양성 중증 근무력증보다 MuSK항체 관련 중증근무력증에서 리툭시맙 치료 이후 더 높은 확률로 병의 완화를 보였다[13].
결론적으로 자세한 병력청취와 신경계진찰을 바탕으로 빠른 MuSK항체 관련 중증근무력증 진단을 바탕으로(Fig.) 효율적인 치료적 선택을 하는 것이 중요하다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print