Creutzfeldt-Jakob병(Creutzfeldt-Jakob disease)은 비정상적인 변형 프라이온으로 인하여 빠르게 진행하는 뇌기능장애를 발생시키는 감염질환이자, 신경퇴행질환이다. 발생요인에 따라 산발(sporadic) Creutzfeldt-Jakob병, 가족/유전(familial/genetic) Creutzfeldt-Jakob병, 획득(acquired) Creutzfeldt-Jakob병으로 분류한다[1,2]. 이 중 산발 Creutzfeldt-Jakob병이 85% 비율로 가장 많이 발생하고, 가족/유전 Creutzfeldt-Jakob병이 10-15%를 차지한다. 또한 변종(variant) Creutzfeldt-Jakob병 및 의인(iatrogenic) Creutzfeldt-Jakob병과 같은 획득 Creutzfeldt-Jakob병이 드물게 발생한다[3]. 국내의 경우 질병관리청에 따르면 2016년부터 2019년 사이 확진 환자 3명, 추정 환자 179명, 의심 환자 6명으로 총 188명의 Creutzfeldt-Jakob병 환자가 보고되었다. 이를 발생요인에 따른 분류 시 산발 Creutzfeldt-Jakob병은 165명(87.8%), 가족/유전 Creutzfeldt-Jakob병은 23명(12.2%), 해당 기간 동안에는 획득 Creutzfeldt-Jakob병은 보고되지 않아 발생 비율은 해외와 유사한 분포를 보이고 있었다[2].
현재까지 가족/유전 Creutzfeldt-Jakob병과 관련된 prion protein 유전자(PRNP gene)의 돌연변이 유형은 약 50개로 알려져 있으며, 흔하게 확인되는 PRNP유전자의 돌연변이는 지역별, 인종별로 다르다[1]. 국내의 경우 2016년부터 2019년 사이 가족/유전 CreutzfeldtJakob병으로 분류된 23명에서 M232R 유형이 6명으로 가장 많았고, V180I 유형 5명, E200K 유형이 4명으로 그 다음을 차지하고 있었다[2]. 국내에서 주로 확인되는 PRNP유전자의 돌연변이 중 V180I 유형은 60-70대의 고연령에 주로 발생하는 반면 M232R 유형과 E200K 유형은 다양한 연령에서 발생하는 특징이 있다. 또한 E200K 유형은 탈수초말초신경병증과 불면을 동반하기도 한다[4]. 현재까지 E200K변이 Creutzfeldt-Jakob병은 드물지만 국내에서 지속해서 확인되고 있는 상황에서도 이에 대한 자세한 임상 경과와 그 특징에 대한 자료가 드물어 본 증례를 보고하는 바이다.
증 례
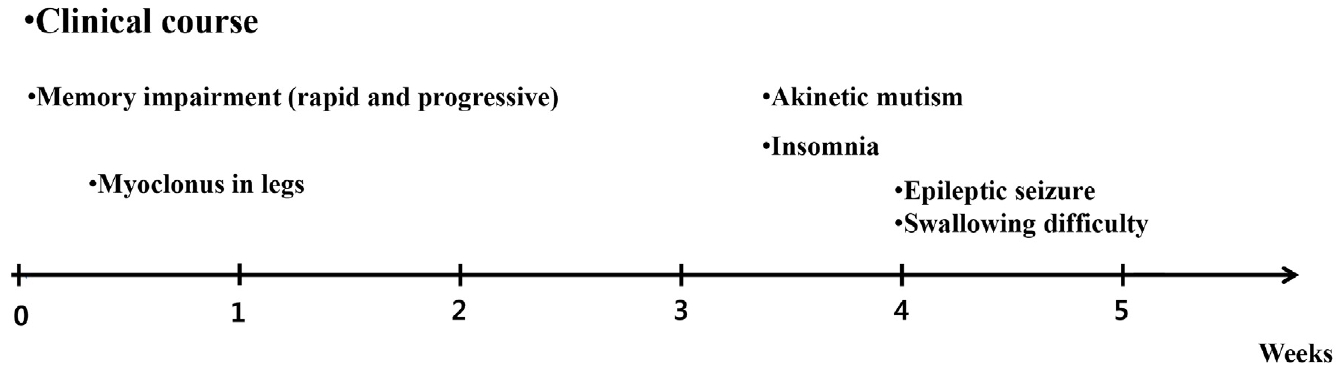
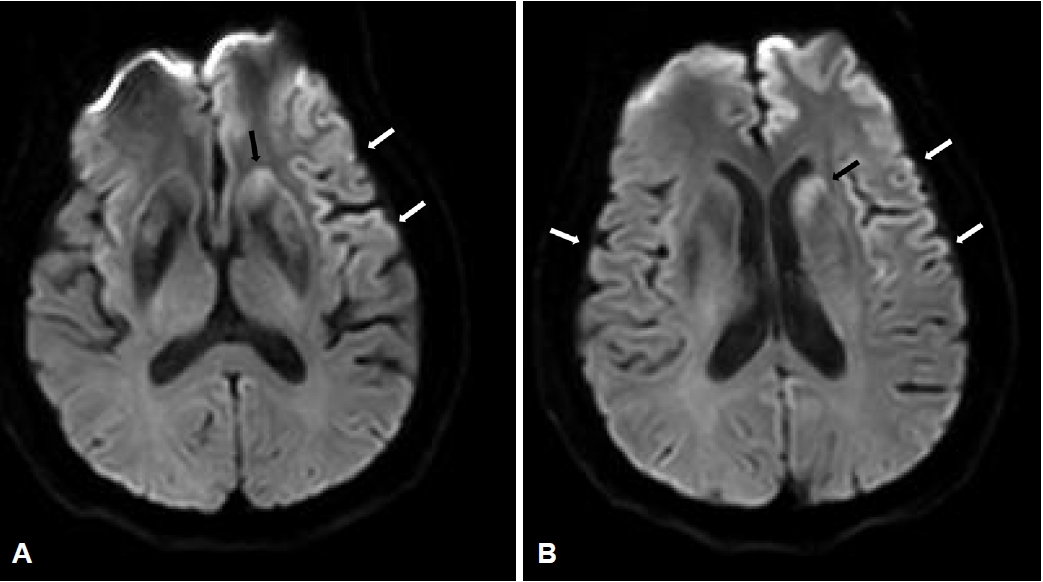
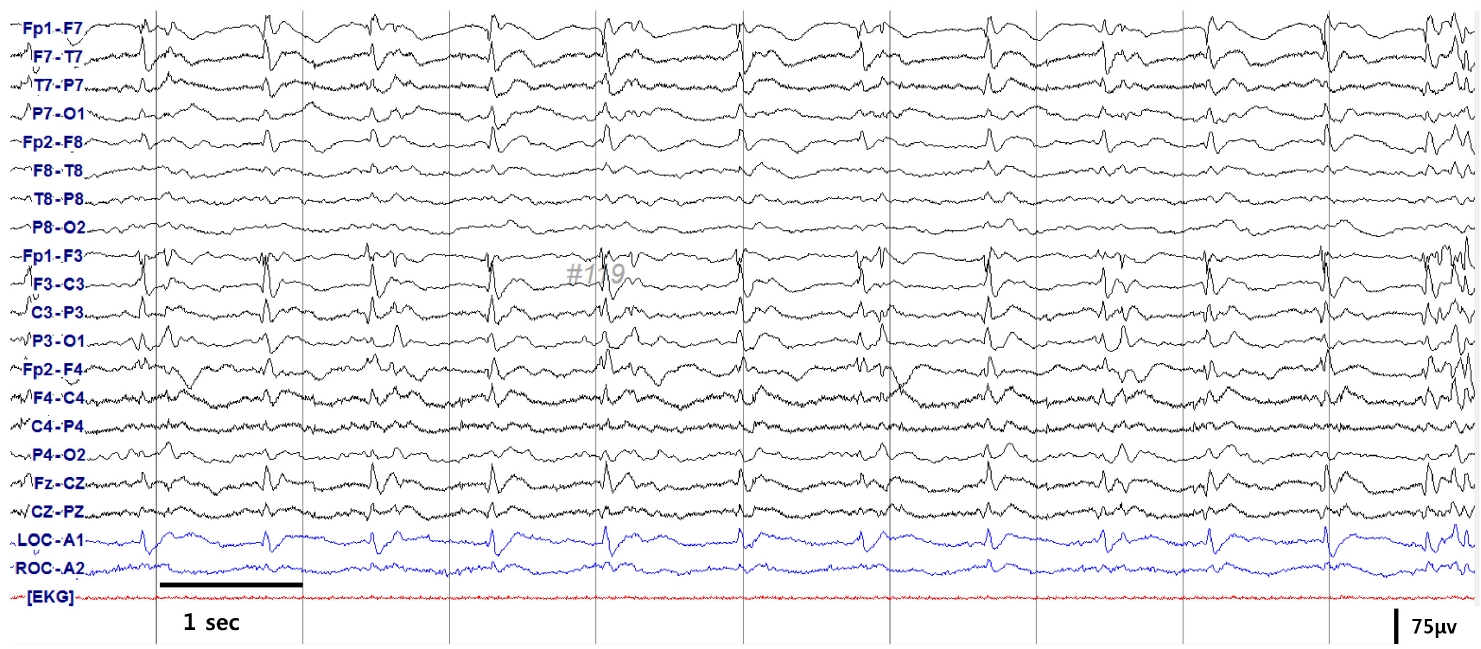
65세 남자가 3주 전부터 갑작스럽게 시작된 기억력저하로 내원하였다. 과거력에서 3년 전부터 불안 및 불면 증상으로 약제를 복용하였으나, 이외 질환으로 복용 중인 약제는 없었다. 또한 신경퇴행질환과 관련된 가족력은 없었다. 가족에 따르면 기억력저하로 직장에서 평상시 하던 일을 처리하는데 문제가 생겼고, 계산을 하는 일에 대해 어려워했다. 3일 뒤부터는 야간 수면 중 양쪽 하지에서 근간대 움직임이 있었다(Fig. 1). 증상은 점차 심해져 일상생활에 장애를 줄 정도가 되어 평소 쉽게 찾아 사용하던 집안의 물건을 찾지 못하여 헤매는 모습이 자주 목격되었다. 혼자서 옷 입기, 식사하기 등도 어려워져 증상 발생 2주 후 타 병원에서 뇌 자기공명영상을 실시하였다. 뇌자기공명영상의 확산강조영상(diffusion-weighted imaging)에서 광범위한 대뇌피질과 꼬리핵에서 고신호강도가 있었다(Fig. 2). 증상 발생 3주 차부터는 대부분의 질문에 본인의 이름 혹은 고향만 반복적으로 말하는 모습을 보여 추가검사를 위하여 본원으로 왔다. 전원 당일 빠르게 진행하는 기억력저하에 대한 감별진단을 위하여 뇌척수액검사 및 뇌파검사를 시행하였다. 뇌파검사에서 주기예파복합체(periodic sharp wave complex)가 보였고(Fig. 3), 뇌척수액검사에서 뇌압은 120 mmH2O, 백혈구는 7/mm3 (참고치 0-5/mm3), 포도당은 61.0 mg/dL (참고치 40-80 mg/dL), 단백은 38.4 mg/dL (참고치 15-45 mg/dL)로 백혈구는 참고치보다 높았으나, 감염질환을 의심할 만큼 의미 있는 상승은 아니었다. 급속진행치매, 근간대경련, 주기예 파복합체 뇌파 소견, 확상강조영상에서 꼬리핵의 고신호강도 병터가 보이고 있어 이를 바탕으로 추정(probable)산발 Creutzfeldt-Jakob병에 해당하여 뇌척수액을 이용한 Creutzfeldt-Jakob병에 대한 검사를 의뢰하였다. 이후 보고된 뇌척수액검사에서 웨스턴블롯분석을 통해 14-3-3단백이 확인되고, RT-QuIC 진단기법을 이용한 검사에서 변형 프라이온단백이 검출되었으며, 타우단백질(15,718.3 pg/mL)도 증가되어 있었다. 또한 PRNP유전자의 중합효소사슬반응(polymerase chain reaction)염기서열분석에서 E200K돌연변이가 발견되었고, 유전자 다형태(polymorphism)는 없었다. 본원 입원 후 의사표현 및 외부 자극에 대한 반응 및 자발적 행동이 점차 줄어드는 무운동무언증을 보이기 시작하였고, 야간 및 주간에 수면을 취하지 않는 불면도 있어 야간에는 쿠에타핀을 25 mg 사용하였다. 기억력저하 발생 후 4주차에는 전신발작을 하였고, 해당 시기에 삼킴곤란을 보이며 흡입 폐렴이 발생하여 비위관 식사를 하게 되었다.
고 찰
본 증례는 급속진행치매, 근간대를 초기 증상으로 하여 뇌 자기공명영상 및 뇌파검사를 바탕으로 추정산발 Creutzfeldt-Jakob병으로 판단하였으나, PRNP유전자검사에서 E200K돌연변이를 확인함으로써 가족/유전 Creutzfeldt-Jakob병으로 최종 진단한 경우이다. E200K돌연변이 확인 이후 신경퇴행질환에 대한 가족력을 재차 확인하였다. 환자의 부모 중 아버지가 80대 중반 치매 증상을 보였다고 기억하고 있었으나, 발생 연령을 고려 시 Creutzfeldt-Jakob병으로 단정 짓기는 어려웠다. 또한 환자는 7남매로 다른 형제자매 및 두 자녀에서도 신경퇴행질환 관련 증상을 보이는 이는 없었다. E200K돌연변이에 대한 환자 가족들 검사는 신경퇴행성질환 관련 증상이 보이기 전까지 시행을 원하지 않아 검사는 하지 않았다.
가족/유전 E200K변이 Creutzfeldt-Jakob병의 임상 특징으로는 질환 초반에 빠르게 진행하는 인지기억저하와 산발 Creutzfeldt-Jakob와 비교하여 상대적으로 낮은 발생 연령, 탈수초말초신경병증을 포함한 말초신경병 및 불면을 들 수 있다[4-6]. 그러나 빠르게 진행하는 인지기능저하는 산발 Creutzfeldt-Jakob병에서도 보이며, 가족/유전 E200K변이 Creutzfeldt-Jakob병 발생 연령은 인종 마다 차이가 있고, 발병 연령이 20대에서 80대 사이에서 넓게 분포하고 있어 임상 증상의 진행속도 및 발병 연령을 통한 산발 Creutzfeldt-Jakob병과의 구별은 매우 어렵다[1,5]. 전원 당시 불면도 본 증례에서 있었으나, 산발 Creutzfeldt-Jakob병 26명, 가족/유전 Creutzfeldt-Jakob병 2명을 포함한 28명의 Creutzfeldt-Jakob병 환자의 수면 특징을 분석한 연구에서 13명(46%)이 불면을 보여 불면도 가족/유전 E200K변이 Creutzfeldt-Jakob병의 특징으로 보기 어렵다[7]. 말초신경병의 동반은 가족/유전 E200K변이 Creutzfeldt-Jakob병의 15%까지 발생하며, 신경집세포(Schwann cell)와 말초신경 역시 변형 프라이온과 관련한 손상을 받기 때문이라고 추정한다[6]. 그러나 본 증례에서는 전원 시 심한 인지기능이 진행된 상태로 말초신경에 대한 검사를 하지는 않아 말초신경병의 동반 여부는 확인하지 못하였으며, 말초신경병증이 가족/유전 E200K변이 Creutzfeldt-Jakob병의 특징인지, 산발 Creutzfeldt-Jakob병 및 가족/유전 Creutzfeldt-Jakob병을 포함한 Creutzfeldt-Jakob병의 특징인지는 추가 확인이 필요하다.
가족/유전 E200K변이 Creutzfeldt-Jakob병은 PRNP유전자의 200번째 유전자부호(codon)에서 글루타메이트가 라이신으로 대체된 변이가 있는 것으로 1989년 폴란드에서 가족/유전 CreutzfeldtJakob병의 변이로 처음 보고되었다[8]. 이후 전 세계에 걸쳐 가족/유전 Creutzfeldt-Jakob병에서 드물지 않게 확인되는 돌연변이 중 하나이다. 지역별로 보면 북미와 남미, 유럽에서 가족/유전 CreutzfeldtJakob병에서 높은 비율을 차지하고, 이와 비교하여 동아시아에서는 상대적으로 낮은 비율을 차지한다[1]. 가족/유전 Creutzfeldt-Jakob병을 포함하여 사람에게 발생하는 프라이온질환은 상염색체우성 형태로 유전되며, 다양한 질환 투과도(penetrance)를 보인다. 본 증례처럼 Creutzfeldt-Jakob병이 의심되어 유전자검사를 통하여 PRNP유전자 변이가 확인된 경우에도 Creutzfeldt-Jakob병의 가족력이 없는 경우가 많으며, 이는 유전자변이별 낮은 질환 투과도 때문으로 생각된다[9]. 따라서 이런 이유 때문에 가족 Creutzfeldt-Jakob병보다는 유전 Creutzfeldt-Jakob병이라는 용어의 사용을 선호하기도 한다[10]. 가족/유전 E200K변이 Creutzfeldt-Jakob병의 30명 사례를 보고한 연구에 따르면 질환 발생 시 30명 모두 산발 Creutzfeldt-Jakob병 형태로 발생하였고, 이후 가계도 및 유전자변이검사를 바탕으로 질환 관련 가족력이 있는 것으로 판단되는 경우는 총 4명 정도만 있었다[5].
본 증례를 통하여 가족/유전 E200K변이 Creutzfeldt-Jakob병은 임상 증상만으로 산발 Creutzfeldt-Jakob병과 구별이 어렵고, PRNP 유전자변이 확인 후 산발 Creutzfeldt-Jakob병과 구별할 수 있음을 확인하였다. 또한 본 증례에서는 평가하지는 못하였으나, 기존 문헌을 통하여 말초신경병도 가족/유전 E200K변이 Creutzfeldt-Jakob병의 동반 증상임을 알 수 있었다. Creutzfeldt-Jakob병은 현재까지 치료법이 없고, 예후가 매우 불량한 질환으로 Creutzfeldt-Jakob병 환자 관리를 통한 의인 Creutzfeldt-Jakob병의 발생과 차단이 중요하다. 산발 Creutzfeldt-Jakob병이 가장 많은 비율을 차지하나, 국내에서도 가족/유전 Creutzfeldt-Jakob병이 드물지만 지속적으로 발생하고 있다. 따라서 Creutzfeldt-Jakob병 환자의 관리 및 감시 측면에서 Creutzfeldt-Jakob병 진단 시 PRNP유전자의 변이 평가 및 확인은 필요하다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print