서 론
다계통위축증(multiple system atrophy, MSA)은 파킨슨증, 소뇌실조, 자율신경이상증, 추체로징후가 특징인 산발신경퇴행질환이다. 주요 증상에 따라 올리브교뇌소뇌위축증(소뇌실조), 줄무늬체흑질변성(파킨슨증), 샤이-드래거증후군(자율신경이상증) 등으로 각기 불려지다가, 1969년에 이들의 증상이 함께 발생할 수 있음을 인지하게 되면서 다계통위축증이라는 하나의 질환개념으로 소개되었다[1]. 이어서 알파시누클레인 단백질로 구성된 은친화 희소돌기아교세포포함소체(argyrophilic oligodendroglial inclusion) 또는 교세포세포질포함소체(glial cytoplasmic inclusion, GCI)가 공통병리 소견으로 밝혀졌다[2]. 임상적으로, 질환 초기에 자율신경이상증상과 함께 우세하게 나타나는 운동 증상이 소뇌실조이면 다계통위축증-소뇌형(MSA-cerebellar type, MSA-C)으로, 파킨슨증이면 다계통위축증-파킨슨형(MSA-parkinsonian type, MSA-P)으로 진단한다[3]. 사후 부검에서 알파시누클레인 양성의 교세포세포질포 함소체가 올리브교뇌소뇌경로(olivopontocerebellar pathway), 줄무늬체흑질경로(striatonigral pathway), 자율신경계, 피질척수로 등 다발신경계에 광범위하게 분포하면 다계통위축증으로 병리확정진단을 내릴 수 있다[2].
대상과 방법
1. 대상
2010년 3월부터 2020년 2월까지 부산대학교병원 뇌은행에 사후 뇌기증을 한 기증자 총 30명 중 병리진단이 다계통위축증인 2명을 대상으로 하였다. 이들의 임상진단은 한 명은 다계통위축증-소뇌형(증례 1)이고, 다른 한 명은 다계통위축증-파킨슨형(증례 2)이다. 자세한 임상 경과는 다음과 같다.
1) 증례 1
62세 여자가 보행장애와 어지럼으로 왔다. 5년 전부터 일어서거나 걸을 때마다 비회전성 어지럼이 반복되어 발생하였다. 혼자 걸을 수는 있었으나 똑바로 걷지 못하고 휘청거렸고, 중심을 잘 잡지못하여 넘어지는 일이 잦았다. 비슷한 무렵부터 요실금이 생겨 수술을 받았고, 이후에도 야간뇨와 빈뇨가 지속되었다. 이러한 증상들은 서서히 악화되었으나, 인지기능은 경미한 기억장애가 동반된 것 외 정상이었다.
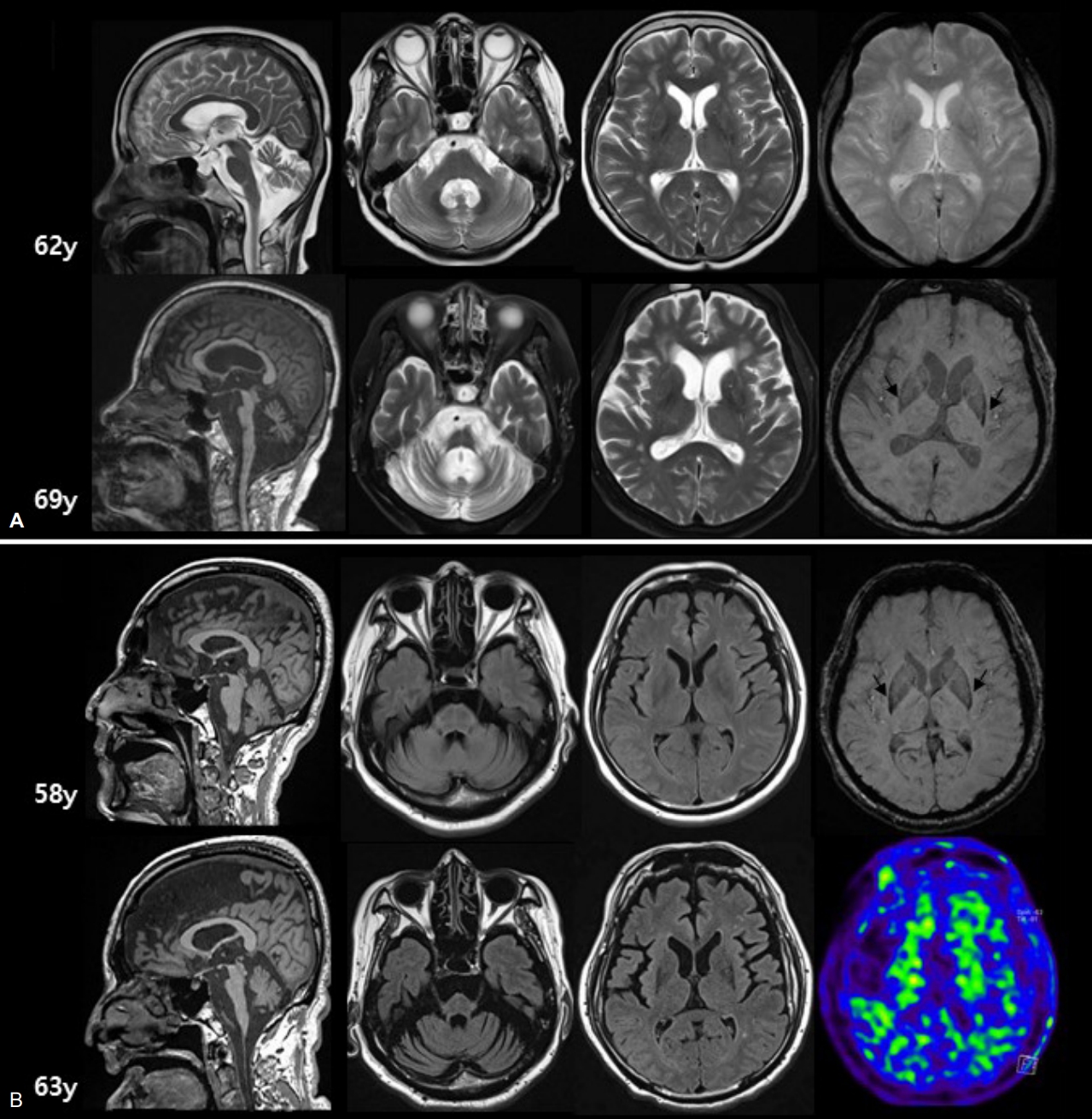
신경계진찰에서 양측 하지 실조와 보행 실조가 있었으나, 양측상지에서 실조나 떨림은 없었고, 우측에서 바뱅스키징후가 양성이었다. 혈액검사에서 특이 소견은 없었다. 서울신경심리선별종합검사(Seoul neuropsychological screening battery, SNSB)에서는 언어기억력에서 경한 기능장애가 관찰되었고, 한국판간이정신상태검사는 30점 만점에 17점(무학, 문맹)이었다. 뇌 magnetic resonance imaging(MRI)에서 소뇌 위축이 확인되었고(Fig. 1-A), 플루데옥시글루코스 F-18 (fluorodeoxyglucose, FDG)양전자단층촬영(positron emission tomography, PET)에서 소뇌 전반에 걸쳐 포도당 대사가 저하되어 있었다. 환자는 5남 6녀 중 넷째로, 어머니와 큰오빠가 환자와 비슷한 증상으로 사망하였다고 보고되어 척수소뇌실조증(spinocerebellar ataxia, SCA) 1형, 2형, 3형, 6형, 7형, 12형 유전자, 치아적핵창백핵루 이체위축증(dentatorubropallidoluysian atrophy, DRPLA) 유전자, 알파시누클레인 유전자(alpha-synuclein gene, SNCA)검사를 진행하였고, 돌연변이는 없었다. 이러한 결과로, 다계통위축증-소뇌형 의심 하에 경과 관찰하였다. 증상 발생 7년째부터 침대에 누워서 생활하게 되었고 기억력을 비롯한 인지장애도 악화되었다. 당시 시행한 한국판간이정신상태검사는 11점이었다. 증상 발생 12년째에는 의미 있는 발화를 할 수 없었고, 양측 상하지는 중력에 반하여 들어올릴 수 없었다. 구음장애와 삼킴곤란이 심하여 피부경유내시경위창냄술을 시행하여 경관식이를 시작하였다. 당시 추적 검사한 뇌 MRI에서 7년 전에 비해 뇌간과 소뇌 위축이 심하게 진행하였다(Fig. 1-A). 이로부터 7개월 후, 증상 발생 12년째, 69세의 나이로 사망하였다.
2) 증례 2
56세 남자가 보행장애와 소변장애를 주소로 병원에 왔다. 3년 전부터 양측 상하지가 뻣뻣해지고 움직임이 느려졌다. 보폭이 좁아지면서 균형을 잡지 못하고 앞으로 넘어지는 일도 여러 번 있었다. 2년 전부터는 표정이 없고 무뚝뚝하다는 말을 듣기 시작하였고 말이 어눌해졌다. 병원에 올 무렵에는 운동완만과 경축이 악화되었고 빈뇨와 발기 부전이 발생하였다. 전립선비대의 병력이 있었고, 신경퇴행질환의 가족력은 없었다. 신경계진찰에서 양측 상하지의 경축과 운동완만, 가면얼굴이 확인되었다. 통합파킨슨병척도 part III는 39점이었고, 파킨슨병 의심 하에 레보도파 복용을 시작하였으나, 이에 대한 반응은 미미하였다. 같은 해 빈뇨와 잔뇨감으로 방광절개술을 시행하였고, 기립 시 심한 어지럼으로 낙상하였다. 낙상 때 둔부에 박힌 이물 제거를 위해 수술하였고, 이후부터 침대에서만 지내게 되었다. 약물에 대한 반응이 없는 상태가 지속되어 내원 후 1년째 레보도파 복용을 중단하였다. 57세에는 심한 삼킴곤란으로 피부경유내시경위창냄술을 시행하여 경관식이를 시작하였다. 이 무렵 통합파킨슨병척도 part III는 73점인 반면, 한국판간이정신상태검사는 30점(학력: 12년)으로 인지기능의 저하는없었다. 58세에 시행한 뇌 MRI에서 소뇌위축과 양측 등가쪽조가비핵에 상자성신호가 확인되었고 다계통위축증-파킨슨형으로 임상진단하였다(Fig. 1-B). 이후 환자는 재활요양병원에 입원하여 지냈고, 증상 발생으로부터 10년 후인 63세에 경과 관찰을 위해 본원에 입원하였다. 세 번째 추적 뇌 MRI에서 소뇌와 뇌줄기의 위축은 심하게 진행하였고, 액체감쇠역전회복(fluid attenuated inversion recovery, FLAIR) 영상에서 등가쪽조가비핵의 신호 강도가 5년 전에 비해 뚜렷하게 낮았다. 18F-N-(3-[18F] fluoropropyl)-2β-carbomethoxy-3β-(4-iodophenyl) nortropane ([18F] FP-CIT)양전자방출단층촬영검사 결과 양측 기저핵에서 도파민운반체 방사선 트레이서의 섭취가 확인되지 않았다(Fig. 1-B). 이로부터 일주일 뒤 말초삽입중심정맥관 시술 중에 갑작스러운 심장마비로 사망하였다.
2. 병리진단과정
뇌적출은 사후 10시간 10분(증례 1)과 14시간 57분(증례 2) 만에 이루어졌다. 적출한 뇌는 반구로 나누어 우측 반구는 관상단면으로 절단 후 -80°C 냉동고에 즉시 저장, 동결하였고, 좌측 반구는 10% 포르말린에 고정하였다. 좌측 반구는 고정 32일(증례 1)과 40일(증례 2) 후에 관상단면으로 절단하였다. 후각망울(olfactory bulb)에서 1-2 mm 앞쪽 지점을 기준으로 먼저 두 부분으로 절단하고, 앞쪽 부분은 그 지점부터 0.8-1 cm 간격으로 균일하게 관상으로 잘랐다. 기준점에서 잘린 뒷부분은 유두체(mammillary body)의 바로 앞쪽, 적핵(red nucleus)과 맞닿는 흑질의 중간부, 중뇌의 등쪽인 덮개(tectum), 띠구조(splenium)의 6-7 mm 앞쪽 부분을 기준으로 차례로 관상면으로 절단하였다. 그리고 남은 부분은 다시 0.8-1 cm 간격으로 균일하게 절단하였다. 이렇게 자른 관상 단면은 질병관리본부 지원 치매뇌조직은행협의체의 파라핀블록절단 공통프로토콜에 따라 총 26부분(전두엽, 측두엽, 두정엽, 후두엽, 기저핵, 시상, 편도, 해마, 중뇌, 뇌교, 연수, 소뇌 등)으로 잘라 파라핀블록을 제작하였다. 완성된 파라핀블록을 8 μm 두께의 얇은 절편으로 잘라 염색하였다. 헤마톡실린-에오진(hematoxylin-eosin, H&E) 염색은 26블록에서 모두 시행하였고, 특정 부분에 한해 다음의 면역조직화학염색을 진행하였다: 베타아밀로이드- MAB5206 단세포군항체(1:300 dilution; Millipore, Burlington, MA, USA), 알파시누 클레인-ab27766 단세포군항체(1:2,000 dilution; Abcam, Cambridge, UK), 인산화타우-CP13항체(1:300 dilution; Peter lab, Manhasset, NY, USA), 인산화TDP-다세포군항체 10782-2AP (1:2,000 dilution;Proteintech, Rosemont, IL, USA)와 T1PPTD-M01 (1:1,000 dilution;Cosmo Bio, Tokyo, Japan).
3. 병리분석방법
줄무늬체흑질과 올리브교뇌소뇌영역의 병리는 기존에 보고된 반정량적분석법을 일부 수정, 적용하여 측정하였다[4,8]. 신경세포소실과 교증(gliosis)은 헤마톡실린-에오진 염색절편으로 평가하였는데, 줄무늬체흑질퇴행(striatonigral degeneration, SND)은 조가비핵(putamen), 창백핵(globus pallidus), 꼬리핵(caudate), 흑질(substantia nigra) 부위를, 올리브교뇌소뇌퇴행(olivopontocerebellar degeneration, OPC)은 아래올리브핵(inferior olivary nucleus), 교뇌(basis pontis), 소뇌를 다음과 같이 네 단계로 구분하였다: 없음(0), 경함(1), 중간(2), 심함(3). 이 중 흑질의 색소신경세포의 소실은 다음과 같은 기준을 적용한다: 없음, 유리 색소도 없음(0); 명확한 신경세포 수의 감소는 없으나, 소량의 유리 색소가 확인됨(1); 명확한 신경세포소실과 유리 색소가 확인됨(2); 신경세포가 거의 없음(3). 알파시누클레인 양성의 교세포세포질포함소체와 신경세포세포질포함소체(neuronal cytoplasmic inclusion, NCI)는 ×20 비율에서 포함소체의 개수를 세어 다음과 같이 네 단계로 구분하였다: 0=포함소체 없음, 1=1-5개의 포함소체, 2=6-19개의 포함소체, 3=20개 이상의 포함소체(Table 1).
4. 병리등급체계
반정량적 분석법으로 얻은 결과를 활용하여 다음과 같이 줄무늬체흑질퇴행과 올리브교뇌소뇌퇴행의 등급을 네 단계로 구분하였다(Table 2).
1) 줄무늬체흑질퇴행(SND)
1단계: 흑질의 신경세포소실은 1 혹은 2이고 창백핵, 꼬리핵 또는 조가비핵은 0 혹은 1. 2단계: 흑질과 조가비핵의 신경세포소실은 2 또는 3이고 꼬리핵과 창백핵은 1 또는 2. 3단계: 흑질과 조가비핵의 신경세포소실은 3.
2) 올리브교뇌소뇌퇴행(OPC)
1단계: 아래올리브핵, 뇌교, 소뇌의 신경세포소실은 0 또는 1, 또는 이 중 한 부위는 신경세포소실이 2이고 나머지 부분에서는 1보다 적음. Torpedo 혹은 empty basket이 조롱박세포층(Purkinje cell layer) 혹은 과립세포층에서 관찰된다. 2단계: 아래올리브핵, 뇌교, 소뇌의 신경세포소실은 2, 또는 이 중 한 부위는 신경세포소실이 3이고 나머지 부분에서는 2보다 적음. Torpedo와 empty basket이 조롱박세포층 혹은 과립세포층에서 많이 관찰된다. 3단계: 아래올리브핵, 뇌교, 소뇌 중 두 개 이상의 부분에서 신경세포 실이 3. 조롱박세포가 거의 관찰되지 않는다.
5. 다계통위축증 병리아형
병리등급체계를 바탕으로 줄무늬체흑질퇴행3단계-올리브교뇌 소뇌퇴행2단계(SND3-OPC2), 줄무늬체흑질퇴행3단계-올리브교뇌 소뇌퇴행1단계(SND3-OPC1), 줄무늬체흑질퇴행2단계-올리브교뇌 소뇌퇴행1단계(SND2-OPC1)는 병리적으로 줄무늬체흑질병리형 다계통위축증(MSA-SND type), 줄무늬체흑질퇴행1단계-올리브교 뇌소뇌퇴행3단계(SND1-OPC3), 줄무늬체흑질퇴행2단계-올리브교 뇌소뇌퇴행3단계(SND2-OPC3), 줄무늬체흑질퇴행1단계-올리브교 뇌소뇌퇴행2단계(SND1-OPC2)는 올리브교뇌소뇌병리형 다계통위축증(MSA-OPC type), 줄무늬체흑질퇴행3단계-올리브교뇌소뇌 퇴행3단계(SND3-OPC3), 줄무늬체흑질퇴행2단계-올리브교뇌소뇌 퇴행2단계(SND2-OPC2)는 줄무늬체흑질/올리브교뇌소뇌 복합병리 다계통위축증(MSA-SND=OPC type)으로 분류하였다(Table 2).
결 과
1) 육안 소견
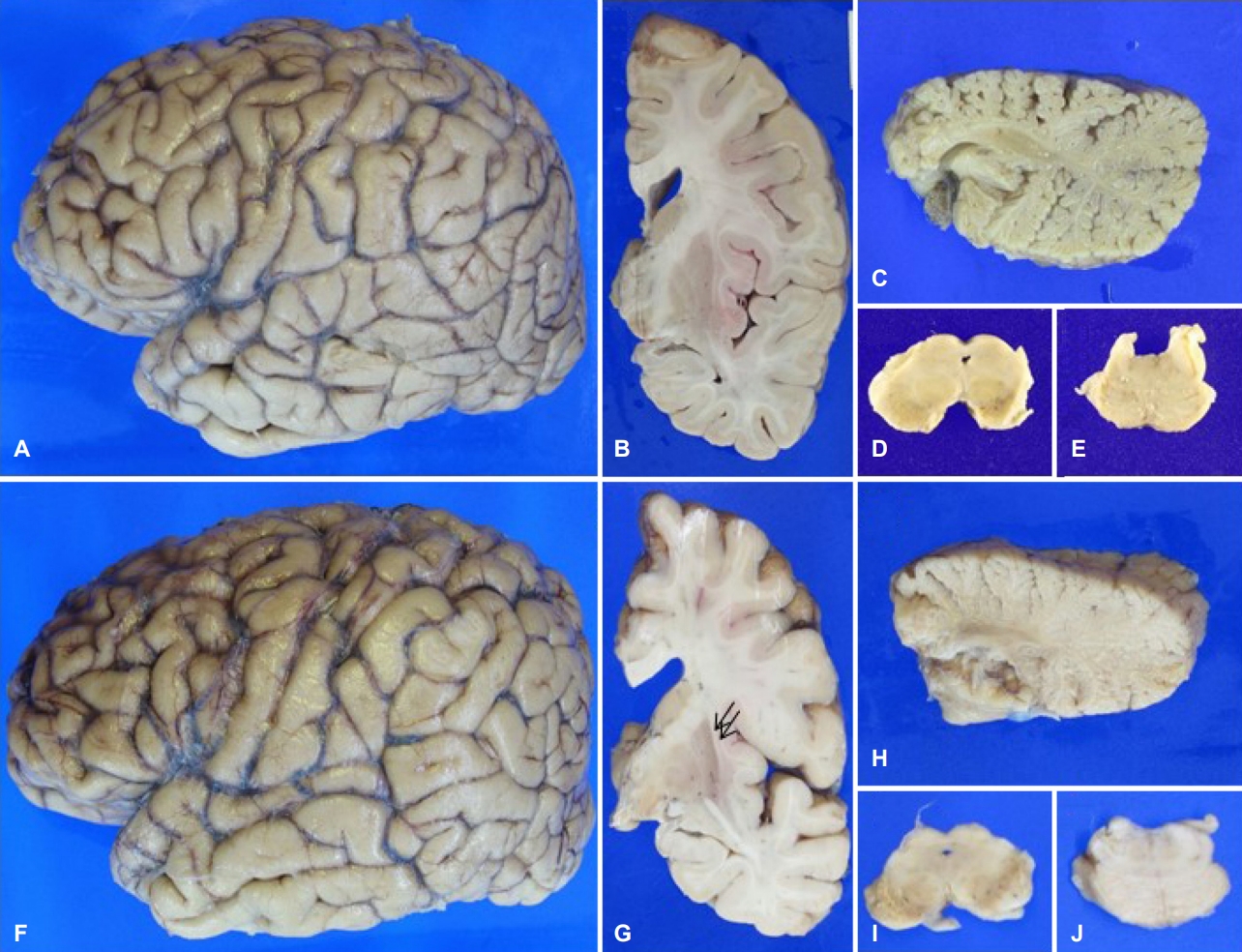
전체 뇌 무게는 증례 1은 1,030 g (좌 456 g, 우 462 g), 증례 2는 1,151 g (좌 521 g, 우 519 g)으로 정상 성인여성(약 1,200 g)과 성인남성(약 1,400 g)의 평균 뇌 무게보다 적었다. 증례 1의 경우 피질 위축은 없었고, 기저핵, 시상, 해마에도 이상 소견은 관찰되지 않았다(Fig. 2-A, B). 하지만 증례 2는 내측 전두엽 부위에 경한 피질 위축이 관찰되고, 후외측조가비핵의 위축이 의심되었다(Fig. 2-F, G). 두 증례 모두 소뇌 피질과 백질의 심한 위축이 확인되고 소뇌 치아핵의 경계가 모호하였다(Fig. 2-C, H). 흑질과 청색반점에서 심한 탈색소 현상이 있었고, 중뇌, 뇌교, 연수등 뇌줄기 전반에 걸쳐 위축이 확인되었다(Fig. 2-D, E, I, J). 뇌혈관이상과 뇌혈관병변은 없었다.
2) 현미경 소견
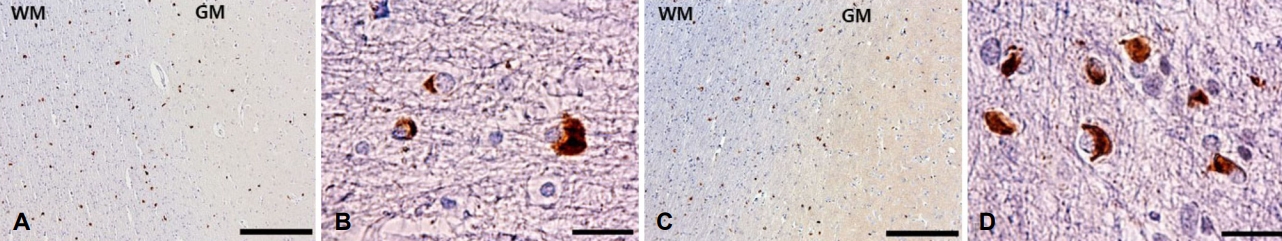
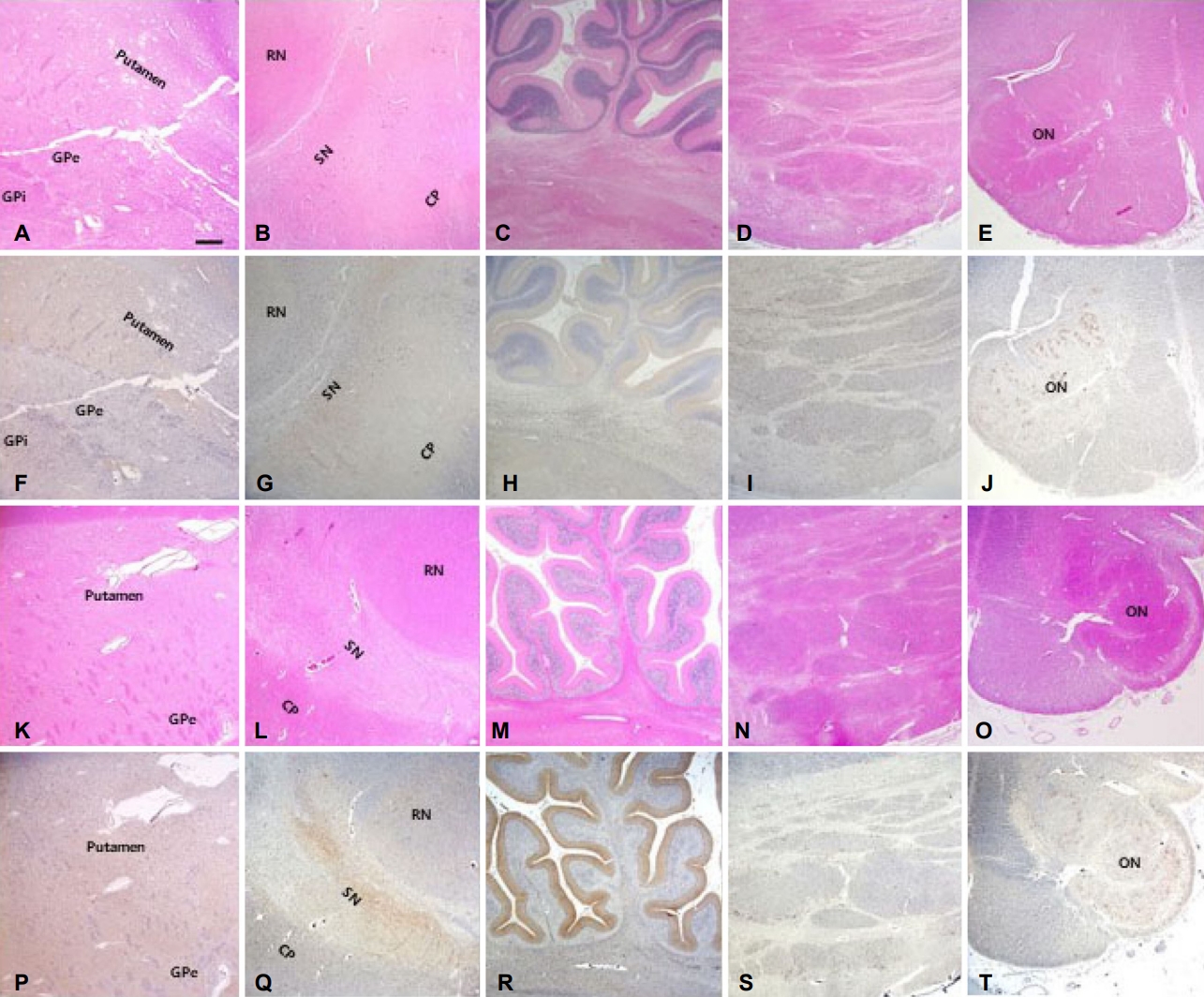
헤마톡실린-에오진 염색에서 대뇌피질의 신경세포소실, 교증, 공포형성(vacuolation) 등의 신경퇴행변화는 확인되지 않았다. 하지만 줄무늬체흑질부와 올리브교뇌소뇌부의 신경퇴행변화는 심하였다. 줄무늬체흑질부의 경우는 파킨슨형인 증례 2에서 소뇌형인 증례 1보다 심하였고, 특히 조가비핵과 흑질의 신경세포소실과 교증이 심한 반면, 창백핵과 미상핵은 상대적으로 신경퇴행변화가 적었다. 올리브교뇌소뇌부의 신경퇴행변화는 두 증례 모두 ‘심함(3)’ 상태였다(Table 2). 알파시누클레인 면역조직화학염색을 하였을 때 전두엽, 측두엽, 두정엽, 편도, 조가비핵, 창백핵, 꼬리핵, 해마, 중심앞이랑, 중심뒤이랑, 새발톱이랑, 소뇌, 중뇌, 뇌교, 연수 등에 알파시누클레인 양성의 교세포세포질포함소체와 신경세포세포질 포함소체가 무수히 많이 관찰되었다(Fig. 3). Perls Prussian blue 염색을 통해, 두 증례 모두 조가비핵에서 철 침착이 확인되었다. 하지만 편도, 흑질, 청반(locus ceruleus) 등운동미주신경핵(dorsal motor nucleus of vagus)에 레비소체는 없었다. 따라서 두 증례 모두 다계통위축증 병리진단 워킹그룹에서 제시한 다계통위축증 병리진단기준에 부합하였다[9]. 이 외에 타우면역조직화학염색에 양성인 신경원섬유매듭이 증례 1의 경우 내후각피질, 해마, 편도, 하측두엽이랑(Braak & Braak 병리분류의 4단계)에, 증례 2의 경우 내후각피질, 해마, 편도(Braak & Braak 병리분류의 3단계)에서 관찰되었다. 베타아밀로이드면역조직화학염색과 transactive response DNA-binding protein 43 (TDP-43) 면역조직화학염색에서 비정상단백질의 침착은 없었다. 이상으로 두 증례 모두 주병리진단은 다계통위축증 그리고 원발연령관련타우병(primary age-related tauopathy, PART)이 동반된 것으로 확정하였다.
2. 다계통위축증 병리아형
증례 1(다계통위축증-소뇌형)과 증례 2(다계통위축증-파킨슨형)의 임상 아형에 따른 병리 소견 차이를 확인하기 위하여 줄무늬체흑질 부분과 올리브뇌교소뇌 부분의 병리를 좀 더 세분화하여 살펴보았다. Table 1에 제시된 바와 같이 올리브뇌교소뇌 부분에서는 두 증례 모두, 신경세포소실과 교증은 심한 상태(3)였고, 알파시누클레인 양성의 교세포세포질포함소체 축적정도는 3(심함), 신경세포세포질포함소체 축적정도는 1(경함)에서 2(중간) 정도로 거의 동일하였다. 하지만 줄무늬체흑질 부분에서는 두 아형 간에 차이가 있었는데, 알파시누클레인 양성의 교세포세포질포함소체와 신경세포세포질포함소체의 축적은 2(중간)에서 3(심함) 정도로 거의 동일하였지만, 증례 2(다계통위축증-파킨슨형)에서 신경세포소실과 교증의 정도가 증례 1(다계통위축증-소뇌형)보다 심하였고, 특히 창백핵과 흑질에서 차이가 있었다(Table 1). 이상의 결과로 증례 1은 SND2 (흑질과 조가비핵의 신경세포소실은 2 또는 3이고 꼬리핵과 창백핵은 1 또는 2)에 부합하였고, OPC3 (아래올리브핵, 뇌교, 소뇌 중 두 개 이상의 부분에서 신경세포소실이 3. 조롱박세포가 거의 관찰되지 않음)에 해당하여 MSA-OPC type으로 분류하였다. 증례 2는 SND3 (흑질과 조가비핵의 신경세포소실은 3), OPC3 (아래올리브핵, 뇌교, 소뇌 중 두 개 이상의 부분에서 신경세포소실이 3. 조롱박세포가 거의 관찰되지 않음)으로 MSA-SND=OPC type으로 구분하였다(Table 2, Fig. 4).
고 찰
다계통위축증은 연간 십만 명당 0.6-0.7명의 발생률을 보이는 드문 산발신경퇴행질환이다. 평균 발생 나이는 56-60세, 평균 유병기간은 5-9년이고 남녀 발생 빈도에 차이는 없다[10,11]. 본 연구의 증례 1과 증례 2는 이러한 다계통위축증의 평균 역학을 따른다. 다만, 증례 1에서 가족력이 보고되어 생전에 척수소뇌실조증과 치아적핵창백핵루이체위축증 관련 유전자검사를 실시하였고, 사후 전장엑솜분석(whole exome sequencing, WES)을 통한 알파시누클레인 유전자(SNCA)검사를 실시하였으나, 돌연변이는 없었다. Wüllner 등12은 보통염색체우성유전 양식을 따르는 가족다계통위축증 독일인 한 가계를 보고하였는데, 최근 구성원의 부검을 통하여 병리적으로도 다계통위축증을 확인하였다.
현재 통용되는 다계통위축증 병리진단기준에 따르면 줄무늬체흑질 혹은 올리브교뇌소뇌 영역에 신경퇴행변화와 관련된 알파시누클레인 양성의 교세포세포질포함소체가 풍부하고 광범위하게 관찰되면 다계통위축증 병리 확정진단을 내릴 수 있다[9]. 우리 증례 1과 증례 2 모두, 이 진단기준에 부합하였고, 원발연령관련타우병이 동반되어 있었으며, 레비소체는 뇌줄기와 편도 등의 호발영역에서 확인되지 않았다.
알파시누클레인 양성의 교세포세포질포함소체가 다계통위축증의 신경퇴행변화를 유발하는 기전은 명확하지 않다. 하지만 교세포세포질포함소체는 신경세포소실 정도와 질환 유병 기간과 상관이 있다[4]. 그리고 질환의 초기상태에서 부검을 하였을 때, 신경세포소실은 흑질과 청색반점 등 일부 영역에만 국한되어 있는 반면, 교세포세포질포함소체는 훨씬 광범위한 영역에 분포하는 것이 보고되어[13], 다계통위축증병리에서 교세포세포질포함소체가 일차적으로 먼저 형성되고, 이어서 이차적으로 신경세포 손상이 유발되는 것으로 여겨진다[14].
병리적으로 줄무늬체흑질 영역에서는 흑질과 등가쪽조가비핵에서 가장 심한 신경퇴행변화(신경세포소실, 교증 등)가 나타나고 미상핵과 창백핵에서는 상대적으로 병리변화가 적다고 보고된다. 올리브교뇌소뇌 영역에서는 소뇌 벌레(vermis) 부위가 가장 심하게 침범되는 것으로 알려져 있다. 본 연구에서는 단 두 증례에 대한 병리 소견만 분석되어 뇌영역별 병리 소견에 대한 통계 분석이 이루어질 수 없었지만, 두 증례 모두에서 흑질과 조가비핵의 신경세포소실과 교증이 창백핵과 미상핵에 비해 정도가 심하여 기존의 보고와 일치하였다[4,8].
파킨슨병과 레비소체병(lewy body disease, LBD)의 주 병리 소견인 레비소체의 동반 정도는 연구마다 차이가 있는데, 서양에서는 10-11%의 다계통위축증에서 레비소체의 동반을 보고하였고, 일본 연구에서는 우리 증례와 같이 레비소체가 동반된 환자가 없었다[4,5,15]. 다계통위축증에 동반되는 병리로는, 우리 증례 1, 2에서 확인된 원발연령관련타우병 외, 알츠하이머병, 진행핵상마비, 은친화입자병(argyrophilic grain disease, AGD), 만성 외상뇌병증(chronic traumatic encephalopathy), aging- related tau astrogliopathy(ARTAG), TDP-43병리 등이 있다[16].
우리 연구에서 임상-병리상관 분석 결과 다계통위축증-소뇌형인 증례 1은 MSA-OPC type으로, 다계통위축증-파킨슨형인 증례 2는 MSA-SND=OPC type으로 진단하였다. Ozawa 등[4]은 100명의 서양인 다계통위축증 환자의 임상-병리 소견을 분석하였는데, 17%에서 MSA-OPC type, 34%에서 MSA-SND type 그리고 가장 많은 49%에서 MSA-SND=OPC type을 확인하였다. 올리브교뇌소뇌병리만 있는 경우(pure OPC type)와 줄무늬체흑질병리만 있는 경우(pure SND type)는 없었다. 이 연구는 다계통위축증의 임상 아형을 소뇌형과 파킨슨형으로 구분하지 않아서 임상 아형과 병리 아형 간의 상관을 정확하게 비교해 볼 수는 없었으나 MSA-SND type은 파킨슨병의 임상진단이 가장 많은 빈도를 차지하였고(50%), MSA-OPC type과 MSA-SND=OPC type은 다계통위축증이 가장 많은 빈도를 차지하였다(각각 76.4%와 73.4%). 물론, 이 다계통위축증이 소뇌형인지 파킨슨형인지 알 수는 없으나, 세 병리 아형 중, MSA-OPC type에서 소뇌증상의 빈도가 가장 높고, MSA-SND type에서 운동완만의 정도가 가장 심하여, 임상 아형과 병리 아형 간의 상관을 충분히 짐작할 수 있었다[4]. 50명의 일본인 다계통위축증 환자의 임상-병리 분석연구는 병리 아형 분포에 있어서 서양인 분석 결과와 차이를 보였다. MSA-OPC type이 40%로, 18%인 MSA-SND type보다 많은 빈도를 차지하였고, MSA-SND=OPC type은 42%로 서양 연구와 마찬가지로 가장 높은 빈도를 보였다[5]. 이러한 동서양의 병리 아형 분포의 차이는 임상 아형 분포에서도 나타난다. 서양에서는 다계통위축증-파킨슨형이 흔한 반면, 일본 환자를 대상으로 한 연구에서는 다계통위축증-소뇌형이 흔하였는데, 이러한 인종 간 차이는 환경, 유전요인에 의한 것으로 추정한다[11,16-19]. 증례 2(다계통위축증-파킨슨형)의 병리 아형인 MSA-SND=OPC type은 초기증상으로 소뇌증상(33%), 파킨슨증상(29%), 자율신경이상증상(29%) 등 다양하게 발현할 수 있는 것으로 보고되었다[4].
Ozawa 등[4]은 줄무늬체흑질 영역에서 흑질의 병리가 다소 적어도 조가비핵의 병리가 동반되면 임상적으로 파킨슨증상이 발현할 수 있고, 이에 비해 올리브뇌교소뇌 영역의 병리가 아주 심한 상태에서도 소뇌증상은 발현이 안될 수 있다고 보고하였다. 본 연구의 증례들은 올리브뇌교소뇌 영역의 병리는 모두 다 심한 상태였다. 뿐만 아니라 증례 2의 경우 뇌영상에서도 올리브뇌교소뇌 영역의 위축이 있었지만, 임상적으로 파킨슨 증상만 두드러졌다. 이러한 임상증상은 흑질과 조가비핵의 심한 신경소실로 인한 결과로 해석된다. 반대로 증례 1의 경우, 조가비핵의 신경세포소실은 심하였으나, 흑질의 신경세포소실은 중간 정도로 확인되어 증례 2와 차이가 있었는데, 이와 같은 병리 소견이 증례 1의 임상 아형(소뇌형)의 배경이 됨을 짐작해 볼 수 있다.
본 연구는 다계통위축증-소뇌형과 파킨슨형으로 발현한 증례들의 부검 소견을 바탕으로 다계통위축증 병리 소견을 확인하고, 기존의 서양 보고를 참고하여 임상증상과의 관련성을 살펴보았다. 비록 각 임상 아형당 한 증례에 불과하지만, 국내 첫 연구로서 의의가 있다. 뿐만 아니라, 임상의들에게 다계통위축증의 병리 소견에 대한 지식을 보급하고, 질환에 대한 이해의 폭을 넓히는 계기가 될 수 있을 것이다. 추후 보다 많은 부검 증례를 통하여 정량화된 결과가 제시될 수 있기를 희망한다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print