축근육의 쇠약으로 발현한 VCP관련봉입소체근육병
VCP-related Inclusion Body Myopathy Presenting with Axial Muscle Weakness
Article information
Valosin-containing protein (VCP)은 ubiquitin-proteasome system을 통한 단백질 분해과정과 자가포식현상(autophagy)에 참여하는 단백질이다[1]. 이를 암호화하는 VCP유전자는 봉입소체근육병(inclusion body myopathy, IBM), 패짓병(Paget disease of bone, PDB), 전두측두엽치매(frontotemporal dementia, FTD)로 구성된 질환인 IBMPFD의 원인유전자로 알려져 있다[1]. IBMPFD에서 세 가지 질환은 다양한 빈도로 나타나는데, 패짓병(43%)과 전두측두엽치매(30%)에 비해 봉입소체근육병의 발현율(90%)이 가장 높다[2]. VCP관련봉입소체근육병은 사지대쇠약(limb-girdle weakness)을 보이는 경우가 가장 많지만, 드물게 먼쪽근육과 축근육의 쇠약을 보이며 어깨종아리쇠약(scapuloperoneal weakness)이 보고되기도 했다[2,3]. VCP 돌연변이는 이외에도 근위축측삭경화증, 유전강직하반신마비, 샤르코-마리-투스병과 같은 다양한 질환과 연관되므로 이들을 VCP관련 다계통단백질병(multisystem proteinopathy)으로 부르기도 한다.
축근육(axial muscle)의 쇠약은 일부 염증근육병과 다양한 유전근육병에서 관찰할 수 있다. 주로 척추옆근의 위축으로 인해 나타나며 심하면 머리처짐증후군(dropped head syndrome)이나 몸통굽힘증(camptocormia)을 보이기도 한다[4]. 사지의 근쇠약이 함께 나타나는 경우가 많지만 축근육의 쇠약만 나타나는 경우도 있다. 얼굴어깨위팔근디스트로피(facioscapulohumeral muscular dystrophy)에서 특히 축근육의 쇠약으로 발현하는 경우가 많다[4].
저자들은 축근육의 쇠약으로 발현한 근육병이 유전자진단을 통해 VCP관련봉입소체근육병으로 진단된 환자의 임상적 특징을 고찰하여 보고하고자 한다.
증 례
60세 여자가 10년 전부터 몸이 뒤로 젖혀진다고 호소하였다. 자세를 유지하기 힘들었고 점차 악화되면서 등과 허리의 통증이 심해졌으며 목을 들기가 어려워졌다. 세면대에서 몸을 숙여 세수를 하는 일도 힘들었는데 상, 하지 쇠약은 동반되지 않았다. 그러나 최근 2년 전부터 다리의 힘이 빠지기 시작하였으며 진료 당시 평지보행은 가능하였으나 계단을 오르기는 어려웠다. 유사한 증상의 가족력은 전혀 없었다.
신경계진찰에서 몸통굽히기가 Medical Research Council (MRC) 등급 4-, 몸통젖히기 2, 허벅지굽히기 4, 허벅지펴기 5-, 다리모으기와 벌리기 5-, 무릎굽히기 4-, 무릎펴기 3, 발등굽히기 3~4로 측정되었으며, 상지의 근육은 5-~5 정도로 확인되었다. 얼굴근육은 정상이었고, 조음장애나 삼킴곤란도 동반되지 않았다. 감각기능과 깊은힘줄반사도 모두 정상이었다. 척추전만자세(lordotic posture)와 뒤뚱걸음(waddling gait)을 보였으며, Gowers 징후가 양성이었다. 신경전도검사는 정상이었고 침근전도검사에서 가쪽넓은근과 장딴지근에서 근육병성 운동단위전위가 두드러지게 관찰되었다. 혈청 크레아틴인산화효소는 265 IU/L (정상, ~208 IU/L)로 약간 상승되어 있었다. 근육 컴퓨터단층촬영에서 양측 중간볼기근(gluteus medius)과 작은볼기근(gluteus minimus), 우측의 가쪽넓은근과 양측 장딴지근이 심하게 위축되었고(Fig. A, B) 요추 자기공명영상촬영에서 척추 옆근도 심하게 위축되었다(Fig. C). 근육병으로 판단하고 우측 위팔두갈래근에서 근생검을 시행하였다. 조직 소견에서 근섬유크기다양성의 증가, 괴사 및 재생, 지방세포의 대체가 광범위하게 관찰되었고 고모리-트라이크롬 염색에서는 일부 근섬유에서 작은 공포가 관찰되었다(Fig. D, E). 근육병패널을 이용한 유전자검사를 시행하였고 VCP유전자에 IBMPFD의 원인으로 보고된 바 있는 c.464G>A (p.Arg155His) 이서형접합돌연변이가 확인되었다(Fig. F). 패짓병에 대한 평가를 위해 측정한 알칼리성인산염분해효소(alkaline phosphatase)는 66-70 IU/L (정상, 35-104 IU/L)였고 뼈스캔에서는 좌측발목의 관절염 소견 외에 다른 이상은 관찰되지 않았다. 전두측두엽 치매에 대한 평가를 시행하고자 하였으나 인지기능 저하가 의심되는 증상이 없었으므로 간이정신상태검사(MMSE, 29/30)를 제외한 다른 검사는 거부하였다.
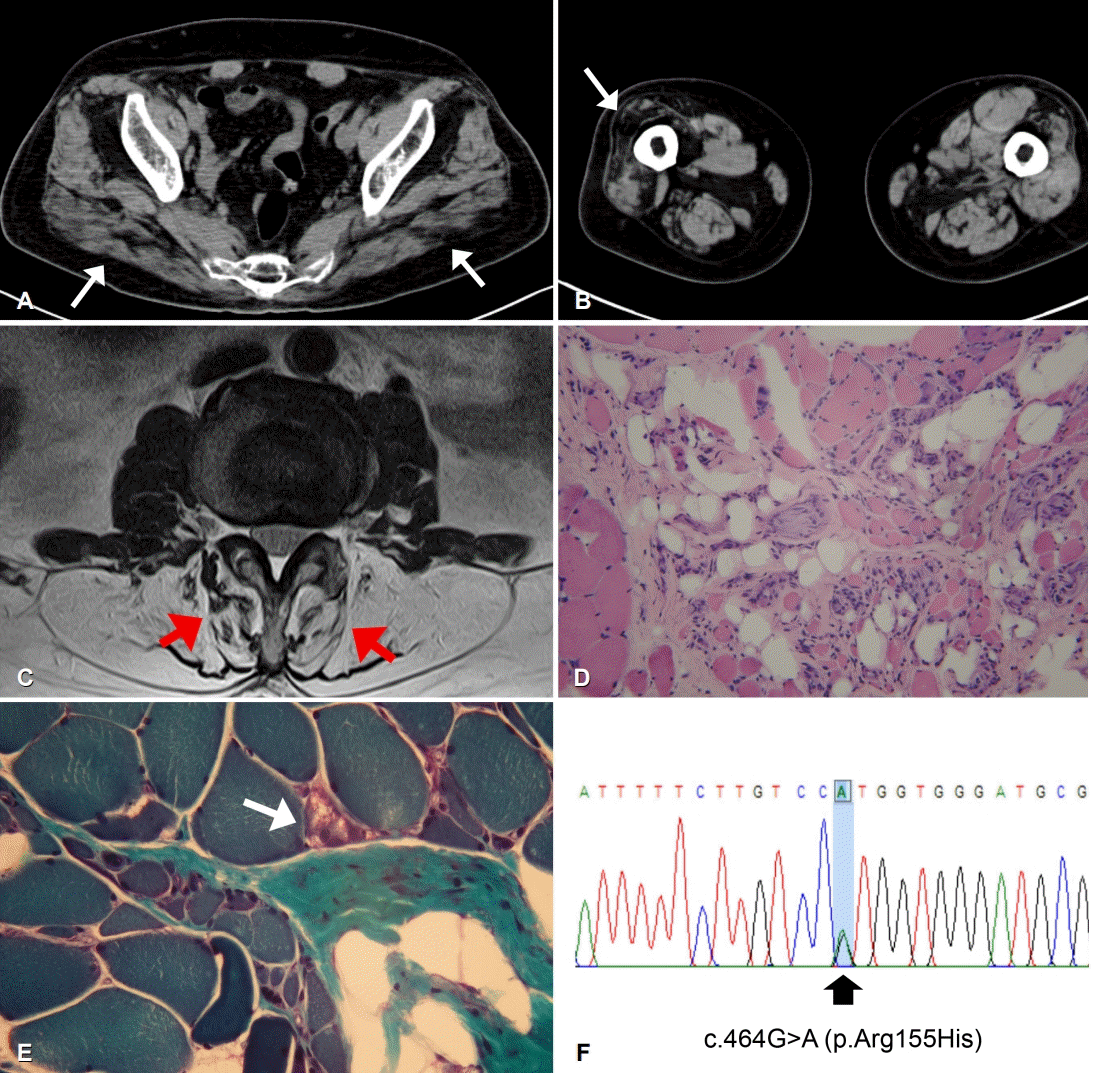
Clinical and pathological features and mutation data of the patient. (A) Muscle computed tomography (CT) showed prominent atrophy of bilateral gluteus medius (arrows) and minimus muscles. (B) Right vastus lateralis (arrow) and posterior thigh muscles were severely atrophied on muscle CT scans. (C) Paraspinal muscles (arrows) were markedly atrophied on magnetic resonance image of lumbar spine. (D) Muscle pathology displayed marked variation in fiber size and extensive fatty infiltration (Hematoxylin and eosin stain, ×200). (E) A few muscle fibers had rimmed vacuoles inside (modified Gomori-Trichrome stain, ×400). (F) Genetic study revealed a heterozygous mutation of c.464G>A (p.Arg155His) in VCP gene.
고 찰
본 증례의 환자에서 확인된 VCP유전자의 c.464G>A (p.Arg155His) 돌연변이는 IBMPFD 환자에서 비교적 자주 발견된다. 그러나 알칼리성인산염분해효소가 정상이었고 뼈스캔에서도 패짓병을 시사하는 소견이 관찰되지 않았다. 자세한 인지기능검사는 시행하지 못하였으나 전두측두엽치매를 시사하는 증상 또한 전혀 없었으므로 증례의 환자는 IBMPFD의 증상 중 유일하게 근육병만을 나타낸 것으로 보인다. 이전의 보고에 따르면 IBMPFD에서의 전두측두엽치매는 조기발현하며 대부분 50대 중반 또는 그 이전에 나타나는 것으로 보인다[1,2,5]. 따라서 앞으로도 이 환자에서 인지기능저하가 발생할 가능성은 비교적 적을 것으로 보이지만 이에 대한 추적 관찰이 필요할 것이다. IBMPFD에서 근육병만을 보이는 경우는 드물지 않다. 관련된 보고에서 30% 이상의 환자가 뼈의 이상이나 인지기능저하를 동반하지 않은 순수 근육병이었으며, 오히려 모든 증상이 나타난 환자는 10%에 불과하였다[2,5]. 다른 증상을 동반하지 않는 VCP관련봉입소체근육병은 임상적, 병리학적 특징이 두드러지지 않으므로 감별진단이 쉽지 않다. 증례와 같이 고모리-트라이크롬염색에서 rimmed vacuole과 같은 공포를 보이는 것이 특징이기는 하나 일부 근세포에서만 관찰되며 그마저도 약 40%의 환자에서만 나타나므로 한계가 있다[5].
본 증례와 같이 상당기간 사지의 근력저하 없이 축근육의 쇠약만 지속된 경우 근육병 진단이 지연될 가능성이 높다. 본 증례의 환자 역시 하지 근력약화가 나타나기 전 8년 동안 요추 문제에 대한 평가만 받았다. 척추옆근의 위약은 몸통젖히기의 근력측정을 통해 평가할 수 있다[4]. 만약 허벅지펴기쇠약이 동반되어 있다면 이로 인해 몸통젖히기에 제한이 생길 수 있으므로 함께 평가하는 것이 중요하다[4]. 척추옆근의 위축은 근육자기공명영상촬영이나 척추자기공명촬영에서 확인할 수 있다. 축근육의 쇠약이 두드러지는 몇 가지 근육병이 있는데, 늦은 나이에 발병하는 축근육병(axial myopathy)의 원인으로 VCP 외에 RYR1유전자가 밝혀진 바 있다[6]. 이 때 조직 소견에서 핵심(core)을 관찰할 수 있다. 얼굴어깨위팔근디스트로피와 함께 뒤셴느근디스트로피(Duchenne muscular dystrophy)에서도 비교적 초기에 척추옆근의 위축이 나타나는 것으로 알려져 있으며 디스펄린병(dysferlinopathy), 칼페인병(calpainopathy)에서도 나타난다[4]. 비전형적인 사례로써 축근육 약화가 발생하기도 하는데, 보통발처짐으로 발현하는 것으로 알려져 있는 GNE근육병에서 심한 척추옆근의 위축으로 발현한 증례가 보고되기도 하였다[7]. 축근육병이 의심될 때 발병연령, 진행경과, 동반되는 임상증상과 조직 소견을 바탕으로 한 감별 진단이 필요하다. 본 증례의 환자는 다른 임상증상이 없었지만 50세 무렵에 처음 증상이 발생하였고 얼굴과 상지의 근육은 침범되지 않았으며 조직검사에서 디스트로핀이나 디스펄린과 같은 단백질발현이 정상적으로 나타났고 rimmed vacuole과 유사한 공포가 관찰되었으므로 VCP관련봉입소체근육병에 부합하는 것으로 보인다.
VCP관련봉입소체근육병과 축근육병은 매우 드문 질환이지만, 축근육의 약화를 시사하는 증상을 보이는 환자에서 적절한 신경계 진찰과 검사를 통해 축근육병을 인지할 필요가 있으며 증례와 같이 늦은 나이에 발병하여 비교적 서서히 진행하는 근력약화에 대해, 뼈의 이상과 전두측두엽치매를 동반하지 않더라도 VCP유전자의 이상을 염두에 둘 필요가 있을 것으로 보인다.